Background: Substantial evidence suggests that amyloid-β (Aβ) species induce oxidative stress and cerebrovascular (CV) dysfunction in Alzheimer's disease (AD), potentia... More
Background: Substantial evidence suggests that amyloid-β (Aβ) species induce oxidative stress and cerebrovascular (CV) dysfunction in Alzheimer's disease (AD), potentially contributing to the progressive dementia of this disease. The upstream molecular pathways governing this process, however, are poorly understood. In this report, we examine the role of heparan sulfate proteoglycans (HSPG) in Aβ-induced vascular smooth muscle cell (VSMC) dysfunction in vitro. Results: Our results demonstrate that pharmacological depletion of HSPG (by enzymatic degradation with active, but not heat-inactivated, heparinase) in primary human cerebral and transformed rat VSMC mitigates Aβ(1-40⁻) and Aβ(1-42⁻)induced oxidative stress. This inhibitory effect is specific for HSPG depletion and does not occur with pharmacological depletion of other glycosaminoglycan (GAG) family members. We also found that Aβ(1-40) (but not Aβ(1-42)) causes a hypercontractile phenotype in transformed rat cerebral VSMC that likely results from a HSPG-mediated augmentation in intracellular Ca(2+) activity, as both Aβ(1-40⁻)induced VSMC hypercontractility and increased Ca(2+) influx are inhibited by pharmacological HSPG depletion. Moreover, chelation of extracellular Ca(2+) with ethylene glycol tetraacetic acid (EGTA) does not prevent the production of Aβ(1-40⁻) or Aβ(1-42⁻)mediated reactive oxygen species (ROS), suggesting that Aβ-induced ROS and VSMC hypercontractility occur through different molecular pathways. Conclusions: Taken together, our data indicate that HSPG are critical mediators of Aβ-induced oxidative stress and Aβ(1-40⁻)induced VSMC dysfunction. Less
Background. Although relaxin causes vasodilatation in systemic arteries, little is known about its role in cerebral arteries. We investigated the expression and role of r... More
Background. Although relaxin causes vasodilatation in systemic arteries, little is known about its role in cerebral arteries. We investigated the expression and role of relaxin in basilar arteries after subarachnoid hemorrhage (SAH) in rabbits. Methods. Microarray analysis with rabbit basilar artery RNA was performed. Messenger RNA expression of relaxin-1 and relaxin/insulin-like family peptide receptor 1 (RXFP1) was investigated with quantitative RT-PCR. RXFP1 expression in the basilar artery was investigated with immunohistochemistry. Relaxin concentrations in cerebrospinal fluid (CSF) and serum were investigated with an enzyme-linked immunosorbent assay. Using human brain vascular smooth muscle cells (HBVSMC) preincubated with relaxin, myosin light chain phosphorylation (MLC) was investigated with immunoblotting after endothelin-1 stimulation. Results. After SAH, RXFP1 mRNA and protein were significantly downregulated on day 3, whereas relaxin-1 mRNA was significantly upregulated on day 7. The relaxin concentration in CSF was significantly elevated on days 5 and 7. Pretreatment with relaxin reduced sustained MLC phosphorylation induced by endothelin-1 in HBVSMC. Conclusion. Upregulation of relaxin and downregulation of RXFP1 after SAH may participate in development of cerebral vasospasm. Downregulation of RXFP1 may induce a functional decrease in relaxin activity during vasospasm. Understanding the role of relaxin may provide further insight into the mechanisms of cerebral vasospasm. Less
Objective: To investigate the changes in the currents of voltage-dependent calcium channels (VDCCs) in smooth muscle cells of basilar artery in a rabbit model of subarach... More
Objective: To investigate the changes in the currents of voltage-dependent calcium channels (VDCCs) in smooth muscle cells of basilar artery in a rabbit model of subarachnoid hemorrhage (SAH). Methods: New Zealand white rabbits were randomly divided into five groups: sham (C), normal (N), 24 hours (S1), 48 hours (S2) and 72 hours (S3) after SAH. Non-heparinized autologous arterial blood (1ml/kg) was injected into the cisterna magna to create SAH after intravenous anesthesia, and 1 ml/kg of saline was injected into cisterna magna in the sham group. Rabbits in group N received no injections. Basilar artery in S1, S2, S3 group were isolated at 24, 48, 72 hours after SAH. Basilar artery in group C was isolated at 72 hours after physiological saline injection. Basilar artery smooth muscle cells were isolated for all groups. Whole-cell patch-clamp technique was utilized to record cell membrane capacitance and VDCCs currents. The VDCCs antagonist nifedipine was added to the bath solution to block the Ca++ channels currents. Results: There were no significant differences in the number of cells isolated, the cell size and membrane capacitance among all the five groups. VDCC currents in the S1–S3 groups had higher amplitudes than those in control and sham groups. The significant change of current amplitude was observed at 72 hours after SAH, which was higher than those of 24 and 48 hours. The VDCCs were shown to expression in human artery smooth muscle cells. Conclusions: The changes of activation characteristics and voltage-current relationship at 72 hours after SAH might be an important event which leads to a series of molecular events in the microenvironment of the basilar artery smooth muscle cells. This may be the key time point for potential therapeutic intervention against subarachnoid hemorrhage. Less
Background. Although relaxin causes vasodilatation in systemic arteries, little is known about its role in cerebral arteries. We investigated the expression and role of r... More
Background. Although relaxin causes vasodilatation in systemic arteries, little is known about its role in cerebral arteries. We investigated the expression and role of relaxin in basilar arteries after subarachnoid hemorrhage (SAH) in rabbits. Methods. Microarray analysis with rabbit basilar artery RNA was performed. Messenger RNA expression of relaxin-1 and relaxin/insulin-like family peptide receptor 1 (RXFP1) was investigated with quantitative RT-PCR. RXFP1 expression in the basilar artery was investigated with immunohistochemistry. Relaxin concentrations in cerebrospinal fluid (CSF) and serum were investigated with an enzyme-linked immunosorbent assay. Using human brain vascular smooth muscle cells (HBVSMC) preincubated with relaxin, myosin light chain phosphorylation (MLC) was investigated with immunoblotting after endothelin-1 stimulation. Results. After SAH, RXFP1 mRNA and protein were significantly downregulated on day 3, whereas relaxin-1 mRNA was significantly upregulated on day 7. The relaxin concentration in CSF was significantly elevated on days 5 and 7. Pretreatment with relaxin reduced sustained MLC phosphorylation induced by endothelin-1 in HBVSMC. Conclusion. Upregulation of relaxin and downregulation of RXFP1 after SAH may participate in development of cerebral vasospasm. Downregulation of RXFP1 may induce a functional decrease in relaxin activity during vasospasm. Understanding the role of relaxin may provide further insight into the mechanisms of cerebral vasospasm. Less
Objective: To investigate the changes in the currents of voltage-dependent calcium channels (VDCCs) in smooth muscle cells of basilar artery in a rabbit model of subarach... More
Objective: To investigate the changes in the currents of voltage-dependent calcium channels (VDCCs) in smooth muscle cells of basilar artery in a rabbit model of subarachnoid hemorrhage (SAH). Methods: New Zealand white rabbits were randomly divided into five groups: sham (C), normal (N), 24 hours (S1), 48 hours (S2) and 72 hours (S3) after SAH. Non-heparinized autologous arterial blood (1ml/kg) was injected into the cisterna magna to create SAH after intravenous anesthesia, and 1 ml/kg of saline was injected into cisterna magna in the sham group. Rabbits in group N received no injections. Basilar artery in S1, S2, S3 group were isolated at 24, 48, 72 hours after SAH. Basilar artery in group C was isolated at 72 hours after physiological saline injection. Basilar artery smooth muscle cells were isolated for all groups. Whole-cell patch-clamp technique was utilized to record cell membrane capacitance and VDCCs currents. The VDCCs antagonist nifedipine was added to the bath solution to block the Ca++ channels currents. Results: There were no significant differences in the number of cells isolated, the cell size and membrane capacitance among all the five groups. VDCC currents in the S1–S3 groups had higher amplitudes than those in control and sham groups. The significant change of current amplitude was observed at 72 hours after SAH, which was higher than those of 24 and 48 hours. The VDCCs were shown to expression in human artery smooth muscle cells. Conclusions: The changes of activation characteristics and voltage-current relationship at 72 hours after SAH might be an important event which leads to a series of molecular events in the microenvironment of the basilar artery smooth muscle cells. This may be the key time point for potential therapeutic intervention against subarachnoid hemorrhage. Less
Background: The study of the cerebrovascular physiology is crucial to understand the pathogenesis of neurological disease and the pharmacokinetic of drugs. Appropriate mo... More
Background: The study of the cerebrovascular physiology is crucial to understand the pathogenesis of neurological disease and the pharmacokinetic of drugs. Appropriate models in vitro often fail to represent in vivo physiology. To address these issues we propose the use of a novel artificial vascular system that closely mimics capillary and venous segments of human cerebrovasculature while also allowing for an extensive control of the experimental variables and their manipulation. Results: Using hollow fiber technology, we modified an existing dynamic artificial model of the blood-brain barrier (BBB) (DIV-capillary) to encompass the distal post-capillary (DIV-venules) segments of the brain circulatory system. This artificial brain vascular system is comprised of a BBB module serially connected to a venule segment. A pump generates a pulsatile flow with arterial pressure feeding the system. The perfusate of the capillary module achieves levels of shear stress, pressure, and flow rate comparable to what observed in situ. Endothelial cell exposure to flow and abluminal astrocytic stimuli allowed for the formation of a highly selective capillary BBB with a trans-endothelial electrical resistance (TEER; >700 ohm cm2) and sucrose permeability (< 1X10-u cm/sec) comparable to in vivo. The venule module, which attempted to reproduce features of the hemodynamic microenvironment of venules, was perfused by media resulting in shear stress and intraluminal pressure levels lower than those found in capillaries. Because of altered cellular and hemodynamic factors, venule segments present a less stringent vascular bed (TEER <250 Ohm cm2; Psucrose > 1X10-4 cm/sec) than that of the BBB. Abluminal human brain vascular smooth muscle cells were used to reproduce the venular abluminal cell composition. Conclusion: The unique characteristics afforded by the DIV-BBB in combination with a venule segment will realistically expand our ability to dissect and study the physiological and functional behavior of distinct segments of the human cerebrovascular network. Less
Von Willebrand factor (vWF), a hemostatic protein normally synthesized and stored by endothelial cells and platelets, has been localized beyond the endothelium in vascula... More
Von Willebrand factor (vWF), a hemostatic protein normally synthesized and stored by endothelial cells and platelets, has been localized beyond the endothelium in vascular disease states. Previous studies have implicated potential non-hemostatic functions of vWF, but signaling mechanisms underlying its effects are currently undefined. We present evidence that vWF breaches the endothelium and is expressed in a transmural distribution pattern in cerebral small vessel disease (SVD). To determine the potential molecular consequences of vWF permeation into the vessel wall, we also tested whether vWF impairs Notch regulation of key smooth muscle marker genes. In a co-culture system using Notch ligand expressing cells to stimulate Notch in A7R5 cells, vWF strongly inhibited both the Notch pathway and the activation of mature smooth muscle gene promoters. Similar repressive effects were observed in primary human cerebral vascular smooth muscle cells. Expression of the intracellular domain of NOTCH3 allowed cells to bypass the inhibitory effects of vWF. Moreover, vWF forms molecular complexes with all four mammalian Notch ectodomains, suggesting a novel function of vWF as an extracellular inhibitor of Notch signaling. In sum, these studies demonstrate vWF in the vessel wall as a common feature of cerebral SVD; furthermore, we provide a plausible mechanism by which non-hemostatic vWF may play a novel role in the promotion of vascular disease. Less
Neprilysin (NEP), which degrades amyloid-β (Aβ), is expressed by neurons and cerebrovascular smooth muscle cells (CVSMCs). NEP immunolabeling is reduced within cerebral... More
Neprilysin (NEP), which degrades amyloid-β (Aβ), is expressed by neurons and cerebrovascular smooth muscle cells (CVSMCs). NEP immunolabeling is reduced within cerebral blood vessels of Alzheimer's disease (AD) patients with cerebral amyloid angiopathy (CAA). We have now measured NEP enzyme activity in leptomeningeal and purified cerebral cortical blood vessel preparations from control and AD patients with and without CAA. Measurements were adjusted for smooth muscle actin (SMA) to control for variations in CVSMC content. NEP activity was reduced in CAA, in both controls and AD. In leptomeningeal vessels, NEP activity was related to APOE genotype, being highest in ε2-positive and lowest in ε4-positive brains. To assess the role of NEP in protecting CVSMCs from Aβ toxicity, we measured cell death in primary human adult CVSMCs exposed to Aβ(1-40) , Aβ(1-42) or Aβ(1-40(Dutch variant)) . Aβ(1-42) was most cytotoxic to CVSMCs. Aβ(1-42) -mediated cell death was increased following siRNA-mediated knockdown or thiorphan-mediated inhibition of NEP activity; conversely Aβ(1-42) -mediated cytotoxicity was reduced by the addition of somatostatin and NEP over-expression following transfection with NEP cDNA. Our findings suggest that NEP protects CVSMCs from Aβ toxicity and protects cerebral blood vessels from the development and complications of CAA. © 2011 The Authors. Brain Pathology © 2011 International Society of Neuropathology. Less
Cerebral amyloid angiopathy (CAA) is an age-associated condition and a common finding in Alzheimer’s disease in which amyloid-β (Aβ) vascular deposits are featured in... More
Cerebral amyloid angiopathy (CAA) is an age-associated condition and a common finding in Alzheimer’s disease in which amyloid-β (Aβ) vascular deposits are featured in >80% of the cases. Familial Aβ variants bearing substitutions at positions 21–23 are primarily associated with CAA, although they manifest with strikingly different clinical phenotypes: cerebral hemorrhage or dementia. The recently reported Piedmont L34V Aβ mutant, located outside the hot spot 21–23, shows a similar hemorrhagic phenotype, albeit less aggressive than the widely studied Dutch E22Q variant. We monitored the apoptotic events occurring after stimulation of human brain microvascular endothelial and smooth muscle cells with nonfibrillar structures of both variants and wild-type Aβ40. Induction of analogous caspase-mediated mitochondrial pathways was elicited by all peptides, although within different time frames and intensity. Activated pathways were susceptible to pharmacological modulation either through direct inhibition of mitochondrial cytochrome c release or by the action of pan- and pathway-specific caspase inhibitors, giving a clear indication of the independent or synergistic engagement of both extrinsic and intrinsic mechanisms. Structural analyses of the Aβ peptides showed that apoptosis preceded fibril formation, correlating with the presence of oligomers and/or protofibrils. The data support the notion that rare genetic mutations constitute unique paradigms to understand the molecular pathogenesis of CAA.—Fossati, S., Cam, J., Meyerson, J., Mezhericher, E., Romero, I. A., Couraud, P. O., Weksler, B. B., Ghiso, J., Rostagno, A. Differential activation of mitochondrial apoptotic pathways by vasculotropic amyloid-β variants in cells composing the cerebral vessel walls. Keywords: cerebral amyloid angiopathy, Alzheimer’s disease, endothelial cells, smooth muscle cells, oligomerization Less
A hallmark of Alzheimer disease (AD) is the deposition of amyloid β (Aβ) in brain parenchyma and cerebral blood vessels, accompanied by cognitive decline. Previously, w... More
A hallmark of Alzheimer disease (AD) is the deposition of amyloid β (Aβ) in brain parenchyma and cerebral blood vessels, accompanied by cognitive decline. Previously, we showed that human apolipoprotein A-I (apoA-I) decreases Aβ(40) aggregation and toxicity. Here we demonstrate that apoA-I in lipidated or non-lipidated form prevents the formation of high molecular weight aggregates of Aβ(42) and decreases Aβ(42) toxicity in primary brain cells. To determine the effects of apoA-I on AD phenotype in vivo, we crossed APP/PS1ΔE9 to apoA-I(KO) mice. Using a Morris water maze, we demonstrate that the deletion of mouse Apoa-I exacerbates memory deficits in APP/PS1ΔE9 mice. Further characterization of APP/PS1ΔE9/apoA-I(KO) mice showed that apoA-I deficiency did not affect amyloid precursor protein processing, soluble Aβ oligomer levels, Aβ plaque load, or levels of insoluble Aβ in brain parenchyma. To examine the effect of Apoa-I deletion on cerebral amyloid angiopathy, we measured insoluble Aβ isolated from cerebral blood vessels. Our data show that in APP/PS1ΔE9/apoA-I(KO) mice, insoluble Aβ(40) is increased more than 10-fold, and Aβ(42) is increased 1.5-fold. The increased levels of deposited amyloid in the vessels of cortices and hippocampi of APP/PS1ΔE9/apoA-I(KO) mice, measured by X-34 staining, confirmed the results. Finally, we demonstrate that lipidated and non-lipidated apoA-I significantly decreased Aβ toxicity against brain vascular smooth muscle cells. We conclude that lack of apoA-I aggravates the memory deficits in APP/PS1ΔE9 mice in parallel to significantly increased cerebral amyloid angiopathy. Less
Brain amyloid-beta (Abeta) peptide accumulation and aggregation are critical events in the pathogenesis of Alzheimer disease. Increasing evidence has demonstrated that LR... More
Brain amyloid-beta (Abeta) peptide accumulation and aggregation are critical events in the pathogenesis of Alzheimer disease. Increasing evidence has demonstrated that LRP1 is involved in Alzheimer disease pathogenesis. The physiological ligands of LRP1, including apoE, play significant roles in the cellular clearance of Abeta. The receptor-associated protein (RAP) is a specialized chaperone for members of the low density lipoprotein receptor family. RAP shares structural and receptor-binding properties with apoE. Here, we show that RAP binds to both Abeta40 and Abeta42 in a concentration-dependent manner and forms complexes with them. Fluorescence-activated cell sorter analysis showed that RAP significantly enhances the cellular internalization of Abeta in different cell types, including brain vascular smooth muscle, neuroblastoma, glioblastoma, and Chinese hamster ovary cells. This effect of RAP was confirmed by fluorescence microscopy and enzyme-linked immunosorbent assay. RAP binds to both LRP1 and heparin; however, the ability of RAP to enhance Abeta cellular uptake was blocked by heparin and heparinase treatment but not by LRP1 deficiency. Furthermore, the effects of RAP were significantly decreased in heparan sulfate proteoglycan-deficient Chinese hamster ovary cells. Our findings reveal that RAP is a novel Abeta-binding protein that promotes cellular internalization of Abeta. Less
Objectives: To explore the role of angiotensin receptors (AT1 and AT2) and human cytomegalovirus (HCMV) infection in atherosclerosis, and to observe effect of HCMV UL83 g... More
Objectives: To explore the role of angiotensin receptors (AT1 and AT2) and human cytomegalovirus (HCMV) infection in atherosclerosis, and to observe effect of HCMV UL83 gene on angiotensin receptor AT receptors. Methods: Cell model of RNA interference (RNAi) HCMV UL83 gene was set up by transfection RNAi into HCMV infected human cerebral artery vascular smooth muscle cells cultured in vitro by application of transfection technique and adoption of RT-PCR, etc. Effect of HCMV on gene expression of AT1 and AT2 receptors in human cerebral artery vascular smooth muscle cells cultured in vitro under the action of Ganciclovir (GCV), AT1 and AT2 receptor antagonists by immunofluorescence technique, RT-PCR and protein electrophoresis. Results: The duplication and protein expression of HCMV UL83 gene mRNA was successfully interfered using RNAi technique. After silencing HCMV UL83 gene, gene expression of AT1 receptor was down-regulated while that of AT2 was up-regulated. AT1 and AT2 receptor antagonists and GCV, under certain condition, could inhibit the activation on AT1 and AT2 receptor in human cerebral artery vascular smooth muscle cells induced by HCMV infection. Conclusion: Cell model of HCMV infected UL83 silence gene was successfully set up in human cerebral artery vascular smooth muscle cells cultured in vitro. HCMV UL83 might play a role by activating AT1 receptor and inhibiting expression of AT2 receptor. Less
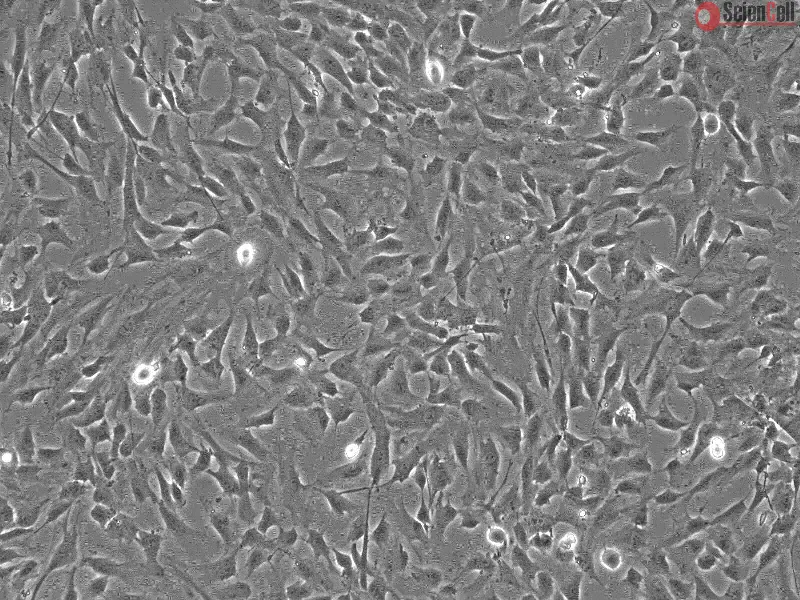
![Human Brain Vascular Smooth Muscle Cells [HBVSMC] - Immunostaining for α-SMA, 400x.](https://sciencellonline.com/media/catalog/product/cache/1/608x608/human-brain-vascular-smooth-muscle-cells.webp)
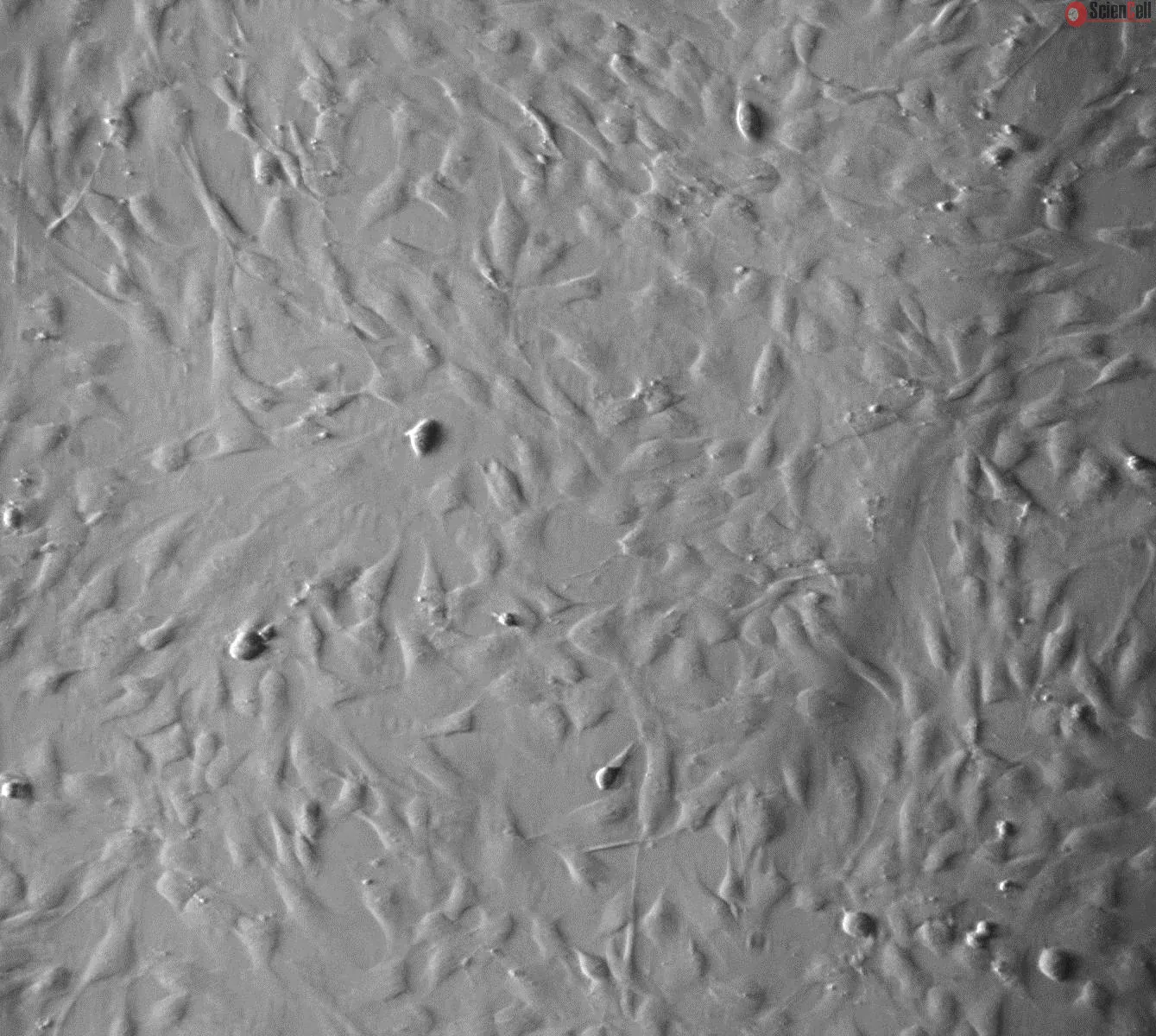
![Human Brain Vascular Smooth Muscle Cells [HBVSMC] - Immunostaining for α-SMA, 400x.](https://sciencellonline.com/media/catalog/product/cache/1/original/human-brain-vascular-smooth-muscle-cells.webp)

,-1-mg-ml--2.jpg)




,-25-ml.webp)